
机器学习 (ML) 目前正在变革蛋白质计算设计的领域,数据驱动的方法在实验成功率上已然超越传统的生物物理方法。然而,现有研究多以个案形式呈现,缺乏系统化和标准化,导致客观比较变得困难。
在近期的研究中,来自德国莱比锡大学的跨学科研究团队在 Rosetta 软件框架的支持下,开发了一个简洁且多样化的工具箱,旨在预测氨基酸的概率,以便于对这些模型进行并行比较。该团队利用现有的蛋白质适应度景观,基于真实的蛋白质设计环境,对新型机器学习方法进行了基准测试。
这一创新方法为研发新药物、如抗体和疫苗,提供了应用潜能,尤其是在应对大流行方面。「我们迫切需要制定此类模型的描述和可用性标准。」该研究的一位负责人,Clara Schoeder 教授指出,「我们的研究为实现这一目标迈出了重要一步。」
研究团队重点关注蛋白质设计中的传统挑战,即采样和评分。他们的研究表明,机器学习方法在剔除采样空间内的有害突变方面具有明显优势。然而,未经优化的评分结果与使用 Rosetta 的评分相比并未显示出显著改进。研究结果表明,目前机器学习的方法更像是对蛋白质设计生物物理方法的补充,而非其替代。
该研究以题为「Self-supervised machine learning methods for protein design improve sampling but not the identification of high-fitness variants」的论文,于2025年2月12日发布在《Science Advances》期刊上。

蛋白质的计算设计与工程一直是科学界的重要目标,旨在快速生成新型蛋白质药物和材料。传统方法如 Rosetta,通过序列突变、结构预测及优化等手段,成功设计了多种蛋白质;而 RosettaScripts 和 PyRosetta 框架则进一步简化了协议的开发过程。尽管如此,蛋白质序列设计仍面临着采样和评分这两大核心挑战。
近年来,机器学习方法在蛋白质结构预测、序列设计及工程方面取得显著突破,诸如 ProteinMPNN 和蛋白质语言模型(PLMs)在纳米材料设计和抗体优化中表现突出。然而,关于 ML 模型是否在整体上优于传统的生物物理设计算法的问题仍然存在,同时复杂的多软件管道也容易引发技术债务及可重复性问题。
为解决这些问题,科学家们将 ESM PLM 家族嵌入 Rosetta,利用 C++ Tensorflow 和 LibTorch 库优化接口,以增强模型的可比性和可移植性。
在此次研究中,莱比锡大学的研究团队评估了新型自监督机器学习方法的有效性,比较其与 Rosetta 等基于生物物理的方法,并探索设计项目的最佳实践。
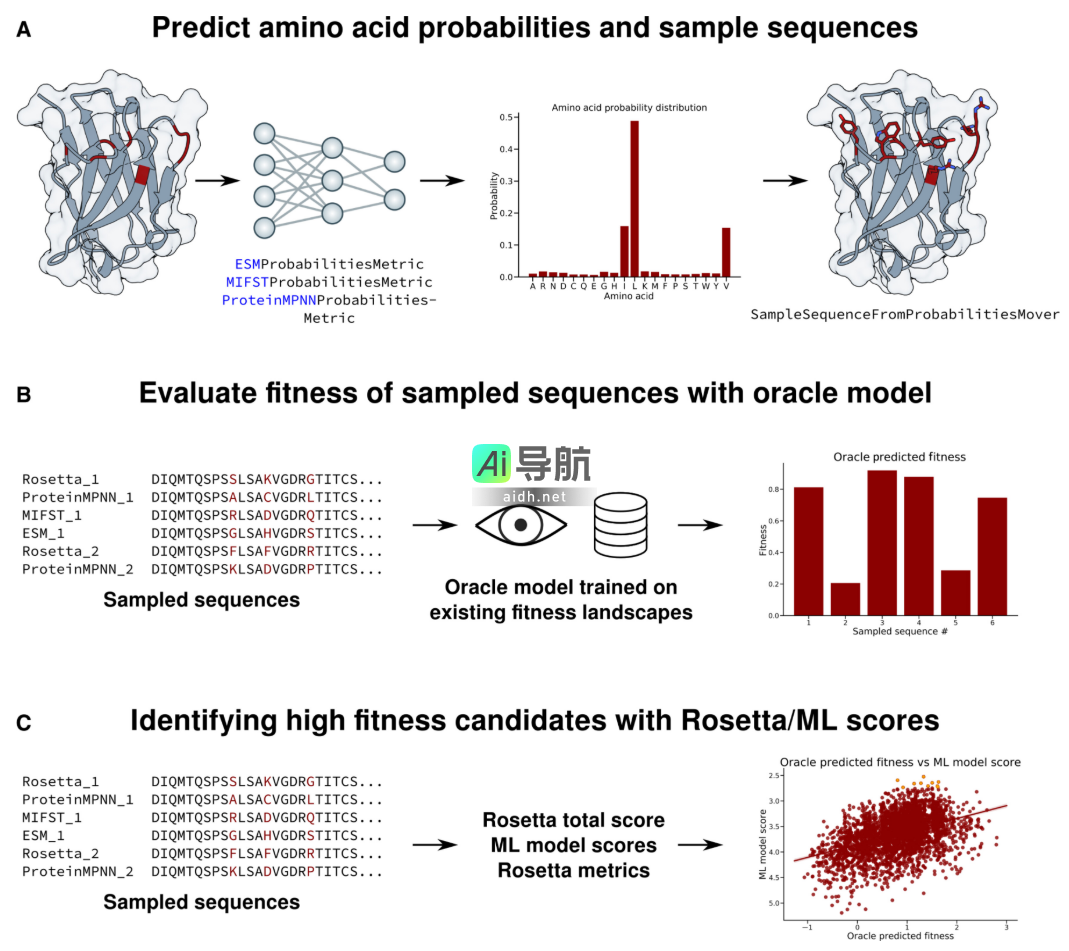
图示:Rosetta 中的机器学习支持框架。(来源:论文)
研究人员利用现有蛋白质适应度景观数据集,对这些工具在常见任务(如提高蛋白质结合亲和力或酶活性)上进行了基准测试,考察它们在零样本方法下的泛化能力。蛋白质工程的两个主要目标分别是生成候选物(即采样突变)和对这些候选物进行评分(即评分突变)。
因此,研究团队在大规模诱变数据集上训练了预测模型(称为「oracle」),以分析16种不同方案下的采样和评分行为。简单而言,尽管机器学习方法在清除序列空间内的有害突变方面表现更佳,但对候选序列的评分和排序仍然是蛋白质设计中的一项挑战。
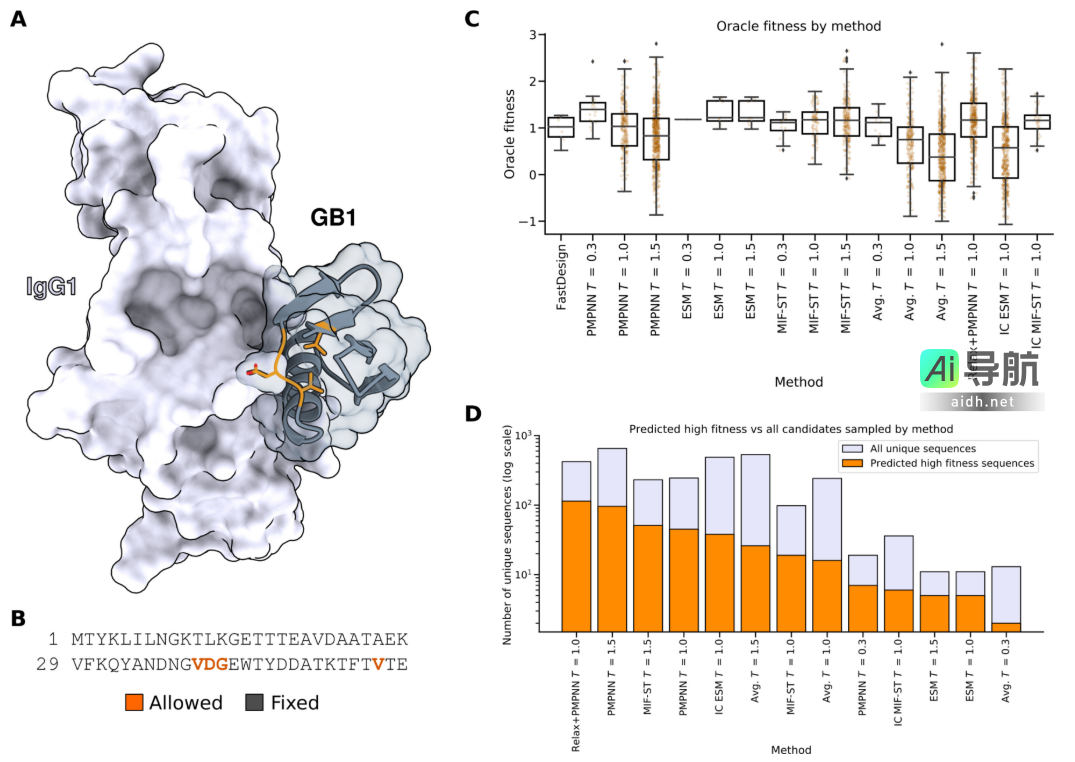
图示:采样突变以提升 GB1 的适应度。(来源:论文)
项目的负责人之一 Jens Meiler 解释道:「我们的研究结果显示,没有单一的人工智能模型或生物物理方法能够完美解决所有设计问题。未来,我们需要谨慎考虑选择哪种模型应对特定的设计任务。我们的研究是提高不同方法间可比性的一次重要尝试。」
具体而言,研究团队探索了自监督机器学习(ML)方法在蛋白质工程中进行序列采样和评分的表现。通过在大型蛋白质适应度数据集上培训 oracle 模型,研究人员发现数据驱动的方法在限制序列空间至无害突变方面明显优于传统方法(如 Rosetta)。
然而,尽管这些方法在采样高适应度序列上表现出色,其预测的适应度值与实际值之间的相关性则相对较低,这使得筛选出最佳候选序列以进行实验验证变得困难。
这一现象表明采样与评分之间存在紧密联系,且评分指标的不完善性可能会对最终结果产生影响。采用更精确的评分函数(例如基于 AF2 的指标)可能在一定程度上缓解这一问题,但在特定复杂案例中(如 emibetuzumab 设计)其效果仍有限。
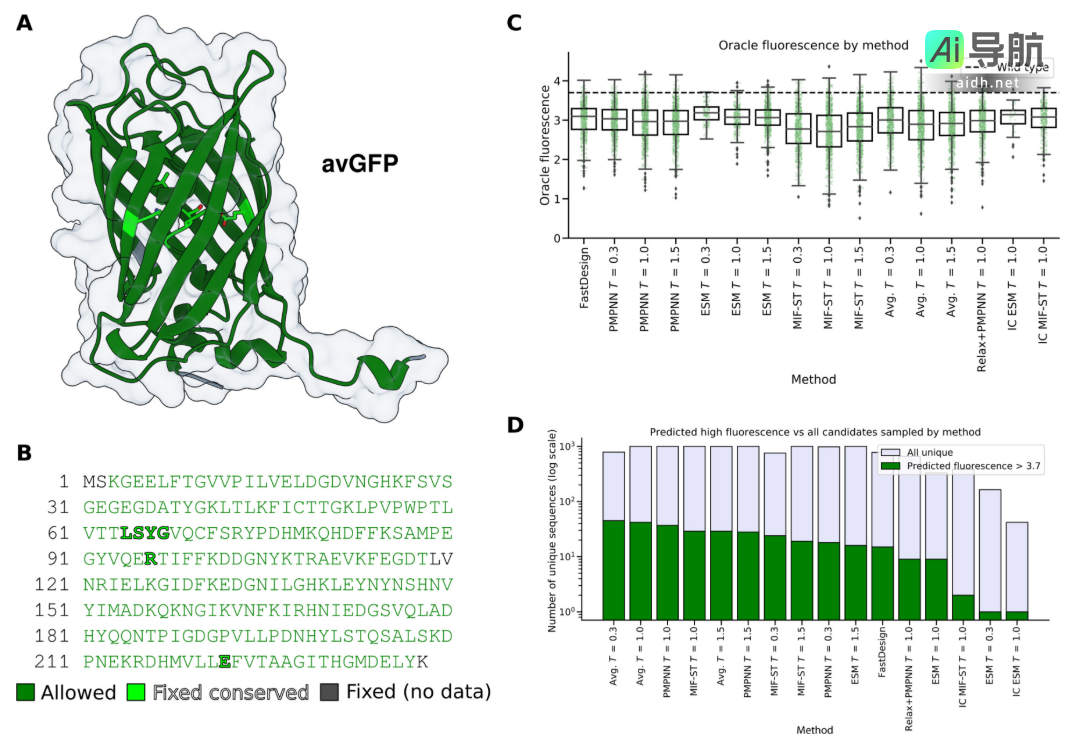
图示:采样突变用于提升 avGFP 荧光。(来源:论文)
研究的最终目标是探索自监督机器学习方法在零样本环境下设计高适应度蛋白质变体的最佳实践。
在复杂功能(例如酶活性)预测方面表现出明显优势,而零样本方法在处理单点突变(如抗体设计)时展现出较佳的效果。例如,ESM-2 模型能够以 49.6% 的准确率预测种系突变。
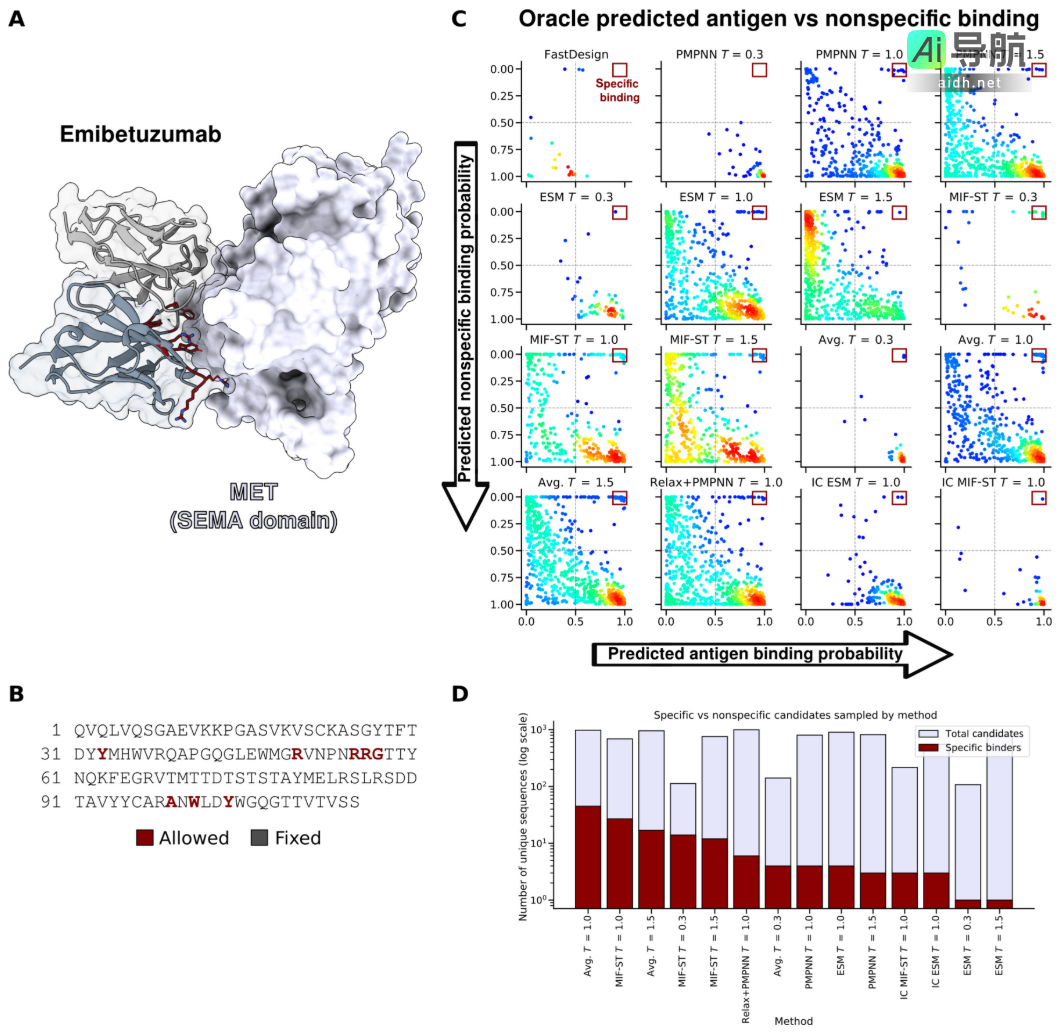
图示:评估埃米贝妥珠单抗双重适应度景观的设计方法。(来源:论文)
Meiler 教授指出:“通过这一进展,我们能够迅速且简便地将人工智能模型与传统方法相结合并并行使用。这大大简化了我们的工作流程,使我们能够充分利用过去 20 年里 Rosetta 所开发的所有基础设施。”
Clara Schoeder 教授补充说道:“我们正在研究哪些方法可以可靠地预测可能导致疫苗候选物的氨基酸变化。”
该研究存在一定的局限性,主要在于对计算机验证的依赖,且所使用的 oracle 模型相对简单,可能无法有效捕捉复杂的突变关系。因此,未来的研究应结合传统方法(如酶工程或抗体设计)进行对比,并探索监督式机器学习模型直接预测蛋白质适应度的潜力。
总体而言,尽管机器学习显著改善了序列采样的效果,但评分和排序仍然面临挑战。研究团队对人工智能与生物物理方法的结合能够提升蛋白质设计效率的前景保持乐观。
论文链接:[Science Advances](https://www.science.org/doi/10.1126/sciadv.adr7338)
相关报道:[Phys.org](https://phys.org/news/2025-02-ai-biophysical-protein.html)
© 版权声明
文章版权归作者所有,未经允许请勿转载。
相关AI热点







暂无评论...