
微软研究院于 2 月 20 日发布博文,正式推出人工智能模型 BioEmu-1。该模型能够预测蛋白质随时间的运动轨迹和形态变化,有望为生物医学、药物发现和结构生物学等领域带来革新。
借助人工智能探索蛋白质
蛋白质在生命活动中扮演着举足轻重的角色,从构成肌肉纤维到抵御疾病,几乎所有生物过程都离不开蛋白质的参与。
近年来,科学家们利用深度学习在蛋白质结构研究方面取得了显著进展,能够根据氨基酸序列精确预测蛋白质结构。然而,仅凭氨基酸序列预测单一蛋白质结构,就好比观看电影中的单帧画面,仅仅呈现了一个高度灵活分子的静态瞬间。
微软 BioEmu-1
与 DeepMind 的 AlphaFold 侧重于确定静态蛋白质结构不同,BioEmu-1 能够模拟蛋白质在不同构象之间的动态转变,从而为理解蛋白质运动规律和设计有效的治疗方案提供了新的工具。
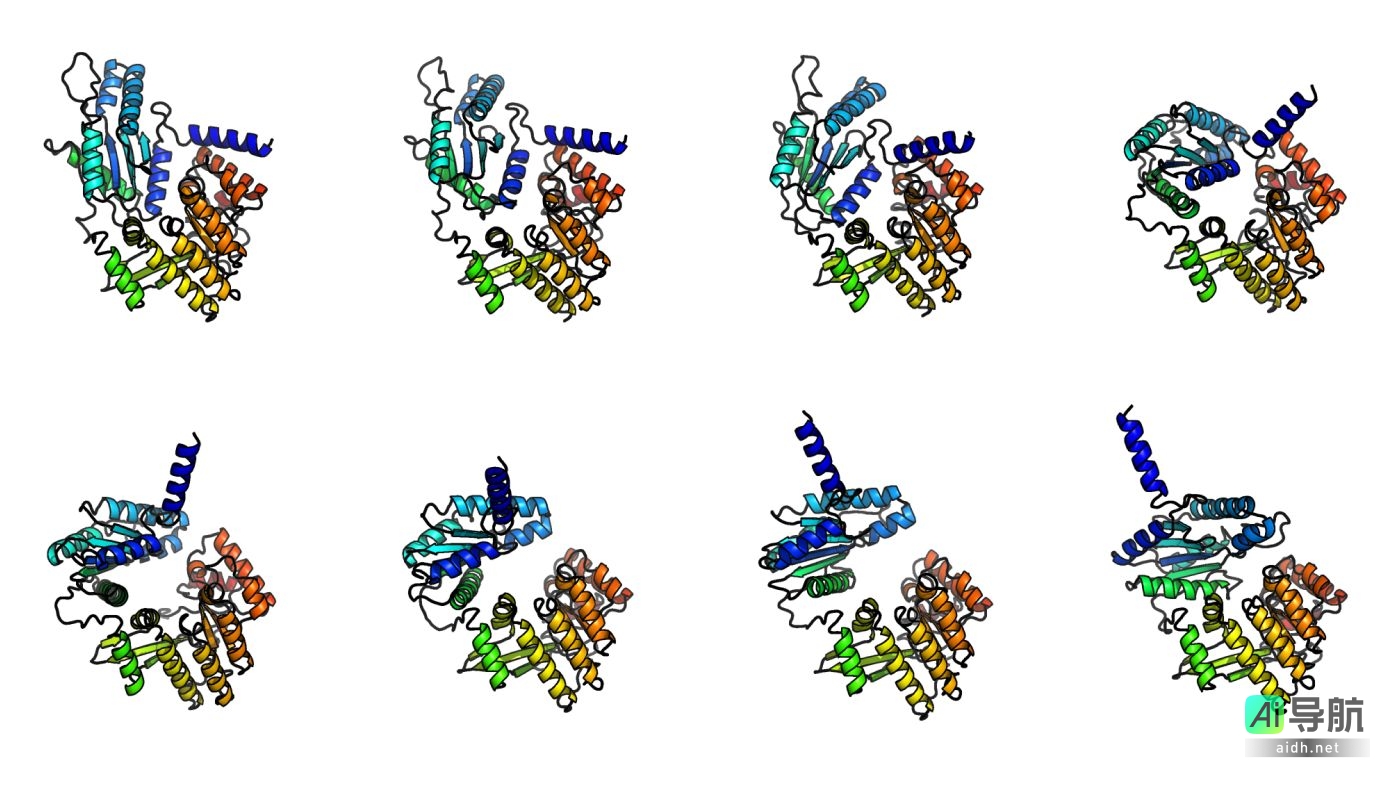
AlphaFold 3 在结构生物学领域取得了重大进展,改进了蛋白质与 DNA、RNA 和小分子之间相互作用的模型,但其局限性在于无法预测蛋白质随时间的动态变化。
BioEmu-1 则弥补了这一不足,它能够生成多个可能的构象,而非单一的最佳拟合结构,这在药物开发领域具有尤为重要的意义。
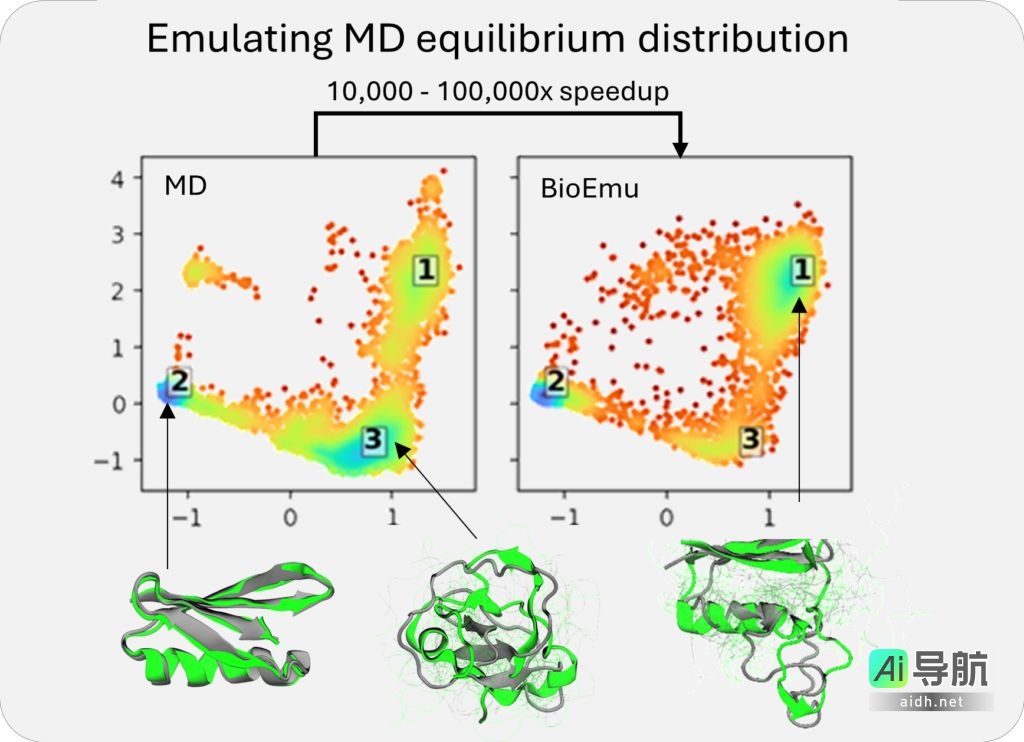
BioEmu-1 采用生成式深度学习方法,通过从大型数据集中学习模式,进而生成与这些模式相符的新样本。该模型结合了静态蛋白质结构、分子动力学模拟数据以及实验稳定性数据进行训练。
BioEmu-1 的核心机制是扩散模型,该模型通过迭代生成蛋白质结构,并根据学习到的约束条件不断提高其准确性。BioEmu-1 的主要输出包括平衡系综的预测和自由能预测。
BioEmu-1 采用了三种类型的数据集进行训练,分别为:(1)AlphaFold 数据库 (AFDB) 结构;(2)广泛的分子动力学 (MD) 模拟数据集;(3)实验性蛋白质折叠稳定性数据集。
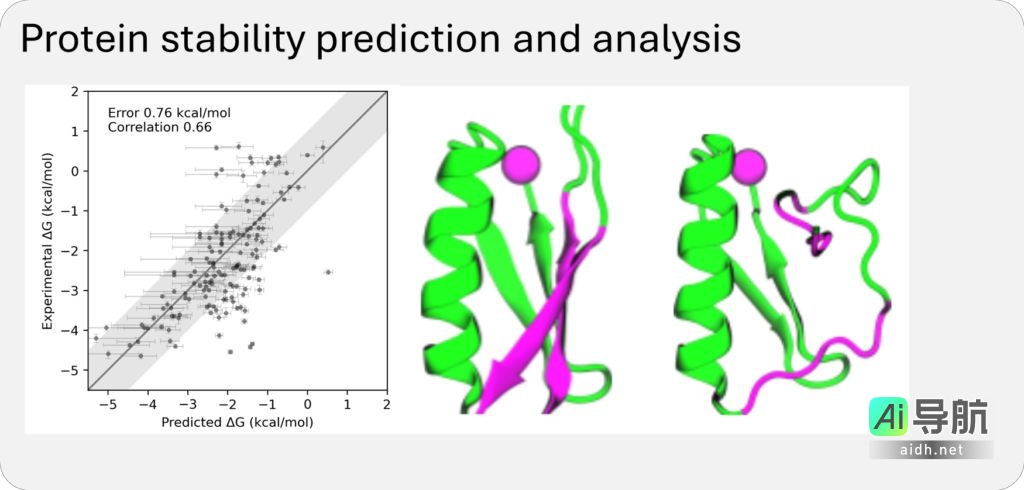
通过对上述数据集的训练,BioEmu-1 能够识别蛋白质序列与多个不同结构之间的映射关系,预测合理的结构变化,并以正确的概率对折叠和未折叠结构进行采样。
BioEmu-1 每小时可以生成数千个蛋白质结构样本,与传统分子动力学模拟需要数周的时间相比,显著加快了研究速度并降低了计算成本。其预测自由能的误差幅度控制在 1 kcal / mol 以内,与传统分子动力学模拟相当,但计算成本却大幅降低。
AI 工具库附上参考地址:
- Exploring the structural changes driving protein function with BioEmu-1
- Microsoft’s New BioEmu-1 AI Model Can Predict How Proteins Move and Change
- Scalable emulation of protein equilibrium ensembles with generative deep learning
© 版权声明
文章版权归作者所有,未经允许请勿转载。
相关AI热点








暂无评论...